
PRINCIPALES SÍNDROMES QUE PUEDEN CAUSAR D.I
:
CUADROS CLÍNICOS ASOCIADOS A LA DISCAPACIDAD INTELECTUAL
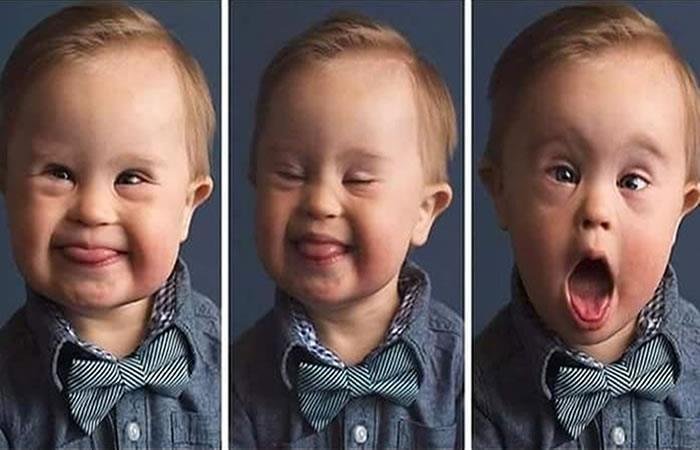
- Síndrome de Down o Trisomía del par 21 :
El síndrome de Down, también conocido como trisomía del par 21, es un trastorno cromosómico provocado por una copia adicional de material genético en el cromosoma 21, que afecta el desarrollo del organismo y del cerebro. Fue descrito por primera vez por el médico inglés John Langdon Down, y en 1959, Jérôme Lejeune descubrió que su causa era una trisomía del par 21.
Su incidencia es de aproximadamente uno por cada 1000 recién nacidos (Roizen & Patterson, 2003) y está influido por la edad materna. Las mujeres de 35 años o más tienen un riesgo significativamente mayor.
Puede ser diagnosticado por medio de un análisis cromosómico prenatal o postnatal, de acuerdo al cual puede ser clasificado en cuatro subtipos: trisomía del par 21, mosaicismo, translocación y duplicación de una porción del cromosoma 21.
Las características clínicas del síndrome de Down son :
• Discapacidad intelectual, generalmente leve; tienen buenas habilidades sociales
• Una apariencia característica, que incluye braquicefalia, pliegues epicánticos, fisuras palpebrales oblicuas hacia arriba, estrabismo, manchas de Brushfield en el iris, nariz pequeña con la base hundida, orejas redondeadas y de baja inserción, macroglosia, boca abierta, cuello corto, braquidactilia, clinodactilia del quinto dedo, huellas dactilares atípicas y separación permanente de los dos primeros dedos de los pies, conocido como el signo de la sandalia
• Retraso ponderal (estatura baja, extremidades cortas y ligamentos laxos)
• A menudo está acompañado de diferentes problemas médicos, entre los que se incluyen cardiopatías congénitas, atresia duodenal, pérdida auditiva, problemas oftalmológicos, hipotiroidismo, demencia de inicio precoz y leucemia.
El síndrome de Down puede ser detectado por medio del cribado prenatal. Los procedimientos de cribado más frecuentes son:
a) la medición de la alfafetoproteína sérica materna (AFP), la gonadotrofina coriónica humana (hCG), el estriol no conjugado y la alfainhibina (INHA) a las 15-20 semanas de gestación
b) la prueba del pliegue nucal por medio de un ultrasonido fetal (translucencia nucal), con una medición de la beta-hCG sérica libre materna y la proteína plasmática A asociada al embarazo a las 10-13.5 semanas de gestación;
c) tanto (a) y (b). En las familias en las que hay un alto riesgo de tener un niño con síndrome de Down, puede ser más adecuado realizar una prueba diagnóstica más invasiva y más precisa en el primer semestre o comienzos del segundo, como una amniocentesis, una biopsia del vello coriónico o una muestra percutánea de sangre del cordón umbilical.
- Síndrome X frágil
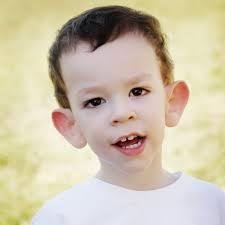
El síndrome X frágil (también conocido como el síndrome Martin-Bell y el síndrome de Escalante) es una enfermedad ligada al cromosoma X, y es una de las formas más frecuentes de discapacidad intelectual hereditaria. También se asocia al trastorno del espectro autista. Martin y Bell describieron por primera vez este trastorno en 1943, y en 1969, Herbert Lubs identificó que estos casos se asociaban a una zona frágil en el cromosoma X.
La mutación que origina el síndrome afecta a una región del cromosoma X donde se encuentra el gen FMR1. El síndrome de X frágil se caracteriza por una expansión de largas secuencias con más de 200 repeticiones del trinucleótido CGG en una región no traducida (SANT) anterior al primer exón 1 del gen FMR1, localizado en la banda q27.3 del brazo largo del cromosoma X (Xq27.3). En personas no afectadas el número de repeticiones en esta región constituye un polimorfismo, siendo habituales valores de entre 5 y 55 repeticiones. La mutación consiste, por tanto, en la amplificación del número de repeticiones del triplete CGG, que silencia la transcripción del gen.
Las manifestaciones clínicas del síndrome de X frágil varían de leves a graves en los aspectos físico, cognitivo, emocional y conductual. Generalmente las mujeres presentan una forma menos grave del trastorno que los hombres. El fenotipo físico incluye una cara larga y estrecha con una frente prominente y orejas protuberantes, hipermovilidad articular asociada a displasia del tejido conectivo, pulgares con doble articulación, pies planos y un macroorquidismo de los varones después de la pubertad. Los individuos con síndrome X frágil a menudo tienen dificultades cognitivas (con un CI que varía desde normal o límite a gravemente bajo), así como problemas en la memoria de trabajo, funciones ejecutivas y en las habilidades matemáticas y visoespaciales. También se evidencia un retraso en el desarrollo del lenguaje en la primera infancia. Trastornos emocionales y del comportamiento son frecuentes, entre ellos trastornos de ansiedad y del estado de ánimo, síntomas del trastorno por déficit de atención con hiperactividad, obsesivos compulsivos (por ejemplo, acciones repetitivas o frases), conductas agresivas y autolesivas, y un temperamento difícil. El síndrome de X frágil es una causa frecuente de trastornos del espectro del autismo, y a menudo se observan también problemas neurológicos, como convulsiones. Además, se cree que las personas que presentan una premutación (cuando el número de repeticiones de CGG oscila entre 55 y 200) tienen un trastorno clínico caracterizado por dificultades de aprendizaje leves, problemas emocionales, insuficiencia ovárica prematura y, en las personas mayores, un trastorno neurodegenerativo llamado síndrome de ataxia/temblor asociado al X frágil, que se suele manifestar en la edad adulta.
El diagnóstico del síndrome de X frágil se realiza por medio de pruebas genéticas de las expansiones de repetición CGG en el gen FMR1 mediante PCR y análisis de southern blot, y deben realizarse a todos aquellos que presenten un retraso en el desarrollo, capacidad intelectual en el rango límite, discapacidad intelectual y trastorno del espectro del autismo. Si se encuentra una longitud normal de la repetición CGG, se debe considerar la realización de una secuenciación del gen FMR1 para excluir la posibilidad de deleción (Garber et al, 2008).
Si se detecta una premutación o una mutación completa, se sugiere realizar asesoramiento genético a toda la familia. Aún cuando el asesoramiento genético no puede prevenir el síndrome de X frágil, es fundamental informarles en relación con la reproducción, y comenzar una intervención adecuada durante la primera infancia.
- Fenilcetonuria
La fenilcetonuria (PKU) es un trastorno metabólico autosómico recesivo descubierto por el médico noruego Ivar Asbjørn Følling en 1934. Se produce por una mutación del gen de la enzima fenilalanina hidroxilasa (FAOH) o de los genes que codifican para las enzimas involucradas en la biosíntesis del cofactor tetrahidrobiopterina (BH4), que provoca una disfunción del metabolismo de la fenilalanina y un exceso de ésta última y de sustancias relacionadas en la sangre, el cerebro y la orina. La presencia excesiva y crónica de fenilalanina en el cerebro es tóxica, y puede provocar daño cerebral grave.
La prevalencia de la fenilcetonuria varía ampliamente en todo el mundo por razones étnicas y sociales (p.e., frecuencia de consanguinidad). En Europa, la prevalencia es aproximadamente un caso por cada 10000 nacidos vivos, en Turquía, de uno de cada 4000, en Latinoamérica, uno de cada 25000-50000, y en algunas regiones de China, uno de cada 100000 nacidos vivos (Blau et al, 2010).
Fenilcetonuria se diagnostica si existe una concentración elevada de fenilalanina en el análisis de aminoácidos en sangre (>120 µmol/L). Para determinar si el paciente con hiperfenilalaninemia tiene un déficit en la biosíntesis o en la regeneración de BH4, debe realizarse una medición de las pterinas urinarias o de la dihidropterina reductasa en los glóbulos rojos por medio de una muestra de sangre en filtro de papel, o una prueba de carga de BH4 (Blau et al, 2005). Los patrones de pterinas urinarias pueden diferenciar diversos tipos de PKU:
• PKU clásica con una deficiencia de PAH: las pterinas totales están elevadas, pero la proporción entre la neopterina y la biopterina es normal
• Deficiencia de la GTP ciclohidrolasa I (GTP-CH): las biopterinas totales están muy bajas o no se detectan
• 6-piruvil-tetrahidropterina sintetasa (6-PTS): la neopterina está elevada pero la biopterina está disminuida
• Deficiencia de la pterina-4a-carbinolamina dehidratasa: la neopterina está elevada, mientras que la biopterina está baja o en el límite, y la primapterina está alta
• Deficiencia de la dihidropteridina reductasa (DHPR): la neopterina está en el rango normal y la biopterina está elevada.
Los bebés con PKU no presentan signos del trastorno al nacimiento, pero durante los primeros meses progresivamente van apareciendo dificultades del desarrollo acompañadas a menudo de una piel, cabello y ojos más claros, rash eccematoso, olor “rancio”, dificultades motoras, convulsiones, problemas de conducta y un trastorno del espectro del autismo. Un cribado, diagnóstico e intervención temprana pueden evitar que los individuos con PKU tengan un daño cerebral grave. En los recién nacidos, se realiza una prueba de cribado en sangre para la PKU a los 3 a 7 días después del nacimiento, y se repite aproximadamente dos semanas más tarde para confirmar el resultado. Si el resultado es positivo, se realizan pruebas adicionales.
Tratamiento debe comenzar tan pronto como se confirme la PKU. La intervención más importante y efectiva es la restricción de fenilalanina en la dieta: leche de fórmula libre de fenilalanina con bajo nivel de proteínas; evitar alimentos ricos en proteína (como la carne, pescado, leche, huevos, pan común, mayoría de los quesos, frutos secos y semillas) y los que contienen aspartamo (harina, soya). Para los pacientes con el tipo BH4-responsivo, identificado por la prueba de carga de BH4, pueden prescribirse preparaciones que contienen BH4. Otros enfoques, como el tratamiento de aminoácidos neutros largos, el uso de la fenilalanina-amonio-liasa y la terapia de genes, aún se encuentran en estudio (Blau et al, 2010). Para mejores resultados, la fenilalanina sérica debe monitorizarse regularmente durante toda la vida, en caso que sea necesario hacer modificaciones en el tratamiento.

- Hipotiroidismo Congénito
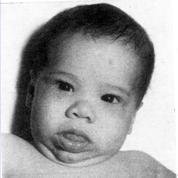
El hipotiroidismo congénito es una enfermedad endocrina provocada por una deficiencia de la hormona tiroidea tras el nacimiento, y puede ser permanente o transitorio. El hipotiroidismo congénito permanente generalmente se asocia a una disgenesia tiroidea, a una disfunción de la biosíntesis o metabolismo de la hormona tiroidea, o a una deficiencia de la hormona estimulante de la tiroides (TSH), mientras que el hipotiroidismo congénito transitorio se produce por una deficiencia de yodo, por el consumo materno de fármacos antitiroideos, o por el paso transplacentario de anticuerpos que bloquean los receptores de tirotropina (TRB-Ab). Ocurre en aproximadamente uno de cada 2000 a 4000 nacidos vivos (Rastog et al, 2010).
Se diagnostica midiendo la TSH sérica y la T4 libre o la T4 total, en conjunto con la captación de T3 con resina. De acuerdo a los rangos de referencia normativos por edad, se confirma un hipotiroidismo congénito primario si la TSH está elevada y la T4 libre o la T4 está disminuida o normal, y se confirma un hipotiroidismo congénito secundario (central) si la T4 está baja pero la TSH no está elevada. Existen otras pruebas diagnósticas para determinar la etiología, entre las que se incluyen medir el yodo urinario, la captación y gammagrafía con radionucleidos, ultrasonografía de la tiroides, medición de la tiroglobulina sérica, determinación de la presencia de anticuerpos antitiroideos, evaluación de la deficiencia de otras hormonas pituitarias, MRI cerebral, y pruebas genéticas.
Las características clínicas del hipotiroidismo congénito en los infantes son: ictericia persistente, disminución de la ingesta, excesiva tranquilidad, dormir en exceso, constipación, baja temperatura corporal, llanto atípico, hernia umbilical, bradicardia, hipotonía y reflejos retardados. Algunos tienen un bocio palpable. La apariencia física incluye una fontanela posterior amplia, cara hinchada, nariz aplanada, ojos con pseudohipertelorismo y una boca abierta con macroglosia. Sin tratamiento, el hipotiroidismo congénito provoca un estancamiento ponderal, discapacidad intelectual permanente y problemas cardíacos .
En las familias en riesgo de tener un bebé con hipotiroidismo congénito, es necesario considerar el asesoramiento genético y el diagnóstico prenatal.
- Síndrome de Angelman
El síndrome de Angelman es un trastorno genético complejo que se caracteriza por un retraso en el desarrollo, discapacidad intelectual, dificultades importantes en el lenguaje, convulsiones, ataxia, aleteo de manos, y un comportamiento feliz y agitado, con sonrisas y risa frecuentes. Fue descrito por primera vez por Harry Angelman en 1965. Su prevalencia es de aproximadamente uno cada 10.000 a uno cada 20.000 nacidos vivos (Petersen et al,1995; Steffenburg et al,1996).
El síndrome de Angelman es causado por la pérdida de la contribución materna normal en una región del cromosoma 15, la mayoría de las veces por una deleción de un segmento de este cromosoma. El diagnóstico se basa en la combinación de las características clínicas, de los resultados de las pruebas genéticas moleculares y del análisis citogénetico. Hay criterios diagnósticos consensuados del síndrome (Williams, 2006). Un análisis de las impresiones de metilación del ADN específicas de los padres en la región 15q11.2-q13 del cromosoma detecta aproximadamente un 78% de los individuos con síndrome de Angelman; menos del 1% tienen una reorganización visible citogenéticamente. El análisis de la secuencia UBE3A detecta mutaciones en otro 11%. Como consecuencia, las pruebas de genética molecular identifican alteraciones en aproximadamente un 90% de los individuos con síndrome de Angelman (Dagli & Williams, 2011). Actualmente el síndrome no tiene cura y su tratamiento es sintomático (p.e., control de la epilepsia por medio de medicamentos antiepilépticos).
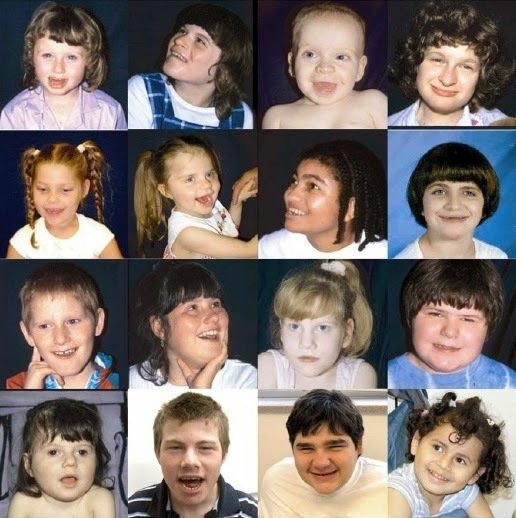
- Galactosemia
La Galactosemia es un trastorno autosómico recesivo de gen único que se asocia a una disfunción de las enzimas que convierten la galactosa en glucosa, lo que resulta en una acumulación de cantidades tóxicas de galactosa en la sangre y los tejidos, provocando discapacidad intelectual y daño en múltiples órganos. Fue descrito por primera vez por Goppert en 1917, y en 1956, Herman Kalckar identificó que se trataba de un defecto en el metabolismo de la galactosa. Su prevalencia es de uno por cada 60.000 nacidos vivos. Según la enzima que se encuentra afectada, la galactosemia se puede clasificar en tres tipos:
• Tipo I, galactosemia clásica, debida a una deficiencia de la galactosa-1- fosfato uridiltransferasa (GALT)
• Tipo II, debida a una deficiencia de la galactokinasa (GLK)
• Tipo III, debida a una deficiencia de la UDP-galactosa epimerasa (GALE).
El diagnóstico de la galactosemia se realiza por medio de una prueba de sangre u orina para detectar la actividad de las tres enzimas mencionadas anteriormente, y para cuantificar los niveles de galactosa. Ahora existen pruebas genéticas moleculares (Elsas, 2010).
Los niños con galactosemia presentan síntomas inespecíficos, como vómito, diarrea, ingesta inadecuada, ictericia prolongada, hepatomegalia, retraso en el crecimiento, letargia y diátesis hemorrágica. Si no es tratada a tiempo, puede ocasionar sepsis, insuficiencia hepática, discapacidad intelectual, retraso en el crecimiento y, finalmente, muerte. Cuando son mayores, niños y adultos pueden presentar complicaciones crónicas o secundarias, incluso los casos con un tratamiento temprano y adecuado, como un retraso en el crecimiento, un funcionamiento intelectual pobre, dificultades en el lenguaje, problemas motores, dificultades de aprendizaje y fallo ovárico.
Para evitar las manifestaciones primarias de la galactosemia, es muy importante realizar pruebas de cribado en todos los recién nacidos y, en los afectados por el trastorno, restringir alimentos y medicamentos que contienen lactosa. Los síntomas mejoran rápidamente y el pronóstico es positivo si la dieta libre de lactosa se comienza en los primeros 3 a 10 días de vida. También es necesario realizar evaluaciones rutinarias de la acumulación de galactosa para ajustar el tratamiento. Otras intervenciones incluyen suplementos de calcio, exámenes oftalmológicos y evaluación del desarrollo y del lenguaje. En familias en riesgo de tener un hijo con el trastorno es recomendable realizar asesoramiento genético y un diagnóstico prenatal.

- Síndrome Alcohólico Fetal
El síndrome alcohólico fetal, la forma más grave de los trastornos del espectro alcohólico fetal, es una causa evitable de la discapacidad intelectual. El síndrome alcohólico fetal se produce cuando la madre consume alcohol durante el embarazo, especialmente durante los primeros tres meses de gestación, lo que provoca un daño importante en el desarrollo del feto, principalmente a nivel cerebral. Prevalencia e incidencia varían según el consumo de alcohol en la población. En Estados Unidos, se estima que 0,2 a 1,5 de cada 1.000 nacidos vivos tiene un síndrome alcohólico fetal (Centros para el Control y Prevención de Enfermedades, 2009).
Muchos profesionales de salud tienen poco conocimiento de este cuadro clínico, y por tanto su detección es baja, y muchos niños afectados no son diagnosticados. Además de los antecedentes de consumo de alcohol de la madre, los profesionales de salud buscan los siguientes signos y síntomas para hacer el diagnóstico (Centros para el Control y la Prevención de Enfermedades, 2009):
• Características faciales anormales
• Problemas del sistema nervioso central
• Problemas de crecimiento.
Los síntomas clínicos varían de acuerdo a la cantidad, frecuencia y duración de la exposición al alcohol, y de las influencias maternas y genéticas. Los bebés y niños con un síndrome alcohólico fetal a menudo presentan un retraso en el desarrollo y una mezcla de anomalías craneofaciales que son el sello distintivo del trastorno, entre las que se incluyen un surco nasolabial sin marcas, un labio superior fino, fisuras palpebrales cortas, pliegues epicánticos, un puente nasal bajo, una nariz corta, malformaciones en las orejas y una mandíbula subdesarrollada. También presentan problemas en el sistema nervioso central, entre los que se incluyen microcefalia, convulsiones, coordinación motora pobre, pérdida auditiva neurosensorial, y dificultades cognitivas y funcionales. Además, el síndrome alcohólico fetal puede predisponer al desarrollo de problemas de salud mental y de consumo de sustancias.
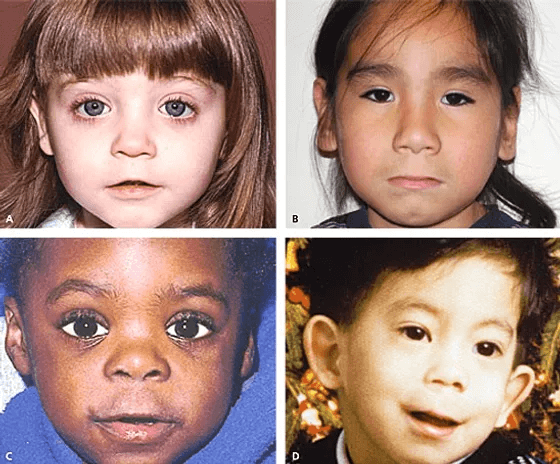
- Síndrome de Williams
Según la asociación Síndrome de Williams de España: “El síndrome de Williams es un trastorno del desarrollo que ocurre en 1 de cada 7.500 recién nacidos”.
Es una enfermedad genética que está caracterizada por:
- Rasgos faciales típicos que pueden no ser evidentes hasta los 2 o 3 años de edad: tienen la frente estrecha y un aumento del tejido alrededor de los ojos, la nariz es corta y antevertida, las mejillas caídas, la mandíbula pequeña, los labios gruesos, presentando asimismo mala oclusión dental.
- Retraso mental leve o moderado con CI medio de 60-70. Existe una asimetría mental, en el sentido de que tienen déficits en algunas áreas (psicomotricidad, integración viso-espacial), mientras que otras facetas están casi preservadas (lenguaje), o incluso más desarrolladas (sentido de musicalidad). Su personalidad es muy amistosa, desinhibida, entusiasta y gregaria.
- Hipercalcemia (niveles de calcio en sangre elevados) en la infancia.
- Estenosis aórtica supravalvular, (un estrechamiento de la arteria principal inmediatamente al salir del corazón).
- Además presentan otras alteraciones; visuales, cardiovasculares, endocrino metabólicas, musculares y esqueléticas, del aparato digestivo, del sistema urinario, etc.

- SínDrome de Prader-Willi
Es una enfermedad congénita, no hereditaria, que puede presentarse en cualquier etnia o sexo.
Se produce por la pérdida o inactivación de genes paternos del cromosoma 15. Los neonatos son hipotónicos, pasivos, con hipoplasia genital y frecuentemente necesitan ser alimentados de forma especial. Durante el embarazo hay una reducción de movimientos fetales.
El diagnóstico en adultos es más fácil si las características clásicas están presentes, ya que el SPW es un conjunto de signos y síntomas que no se manifiestan en todos los afectados, ni aparecen con la misma intensidad o frecuencia.
Incluyen hipotonía muscular, apetito insaciable, obesidad si no se controla la dieta, desarrollo sexual incompleto, retraso en las etapas evolutivas, retraso mental o funcional en diferentes grados, baja estatura, manos y pies pequeños y problemas de comportamiento.
El desarrollo motor está retardado en la mayoría de las etapas evolutivas; por ejemplo caminan alrededor de los 2 años. Presentan el área motora gruesa y el equilibrio afectados; aunque experimentan mejoría siempre evidencian desfase con respecto a los niños de la misma edad.
Tienen problemas de lenguaje, en especial dificultades de articulación. Pero a pesar de que hay un retraso en el desarrollo del lenguaje, la habilidad verbal es frecuentemente buena. La mayoría presenta una deficiencia intelectual de ligera a moderada (en torno a 70). Independientemente de su C.I. presentan problemas de aprendizaje y de lenguaje, por lo que necesitan apoyo en las distintas áreas educativas.
Por lo general, los niños con SPW son extrovertidos, divertidos y generosos. Según van creciendo empiezan a mostrar conductas más problemáticas como son la terquedad, las rabietas, conductas autolesivas y agresividad. La falta de sensación de saciedad es continua y de por vida en las personas afectadas por el SPW.

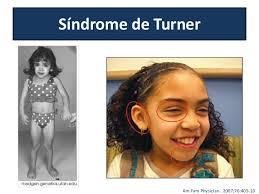
- Síndrome de Turner
Se trata de una monosomía (ausencia de uno de los miembros) en el par 23 y que se da normalmente en las niñas. El síndrome de Turner puede cursar con deficiencia intelectual, aunque no siempre es así, lo normal es que presenten capacidad intelectual normal-baja.
Se caracteriza porque las niñas presentan al nacer manos y pies hinchados, además de un cuello ancho, pero la característica más visible de este síndrome es la baja estatura.
La altura promedio de una mujer adulta con síndrome de Turner es de 1,43 m . La mayoría de las mujeres nace con los ovarios poco desarrollados o sin ovarios. En la mayoría de los casos, la infertilidad resultante no puede corregirse
También son comunes los problemas cardíacos, renales y de tiroides, que deben ser evaluados precozmente.
Alrededor de una de cada diez niñas con síndrome de Turner nace con coartación de la aorta (constricción de la arteria principal que parte del corazón), la cual suele requerir una corrección quirúrgica.
Otras características que se han observado en el síndrome de Turner incluyen problemas de alimentación durante la niñez, infecciones en el oído medio, problemas esqueléticos, piel seca, mandíbula pequeña, presión sanguínea alta, y el desarrollo de diabetes.
- El síndrome de Klinefelter:
Es una cromosomopatía que se caracteriza por la presencia de uno o más cromosomas X adicionales, la forma más frecuente o forma clásica es la 47 XXY. Se trata de la alteración más frecuente de la diferenciación sexual. Fue descrito por primera vez en 1942 por Harry Fitch Klinefelter, Edward Conrad Reinfenstein y Fuller Albright.
Causa una alteración funcional de los testículos en los varones, que se conoce como hipogonadismo. Está presente en aproximadamente el 1% de los niños con discapacidad intelectual y es frecuente entre los niños que acuden a consultas psiquiátricas, o están ingresados en hospitales psiquiátricos; suele acompañarse de un coeficiente intelectual superior a 50. Alrededor de 1 de cada 1.000 nacidos varones tiene un cromosoma X adicional. No se suele diagnosticar hasta la pubertad, siendo hasta entonces los síntomas leves e inespecíficos. Las primeras manifestaciones son de comportamiento, donde los niños en general se muestran ansiosos, inmaduros, agresivos o excesivamente huraños, pudiendo realizar actos antisociales. Los problemas suelen surgir cuando el niño comienza a ir al colegio.
El desarrollo de la pubertad puede estar retrasado. Habitualmente existe un cierto grado de déficit de andrógenos (hormonas sexuales masculinas), aunque algunos pacientes tienen una masculinización normal. Son frecuentes la azoospermía (ausencia de espermatozoides en el esperma) y la esterilidad, aunque se han descrito casos de fertilidad.
Las características que pueden presentar las personas afectadas por este síndrome son:
- Problemas en el habla. Dificultades con el lenguaje expresivo, y por tanto en su habilidad para poner pensamientos y sentimientos en palabras.
- Dificultades de aprendizaje. Retraso en la adquisición de la lectoescritura.
- Bajo nivel de autoestima y timidez.
- Bajo nivel de actividad.
- Problemas de memoria.
- Trastornos de comportamiento.
- Estatura alta.
- Escasez de vello facial y corporal
- Desórdenes autoinmunes.
- Mayor probabilidad de tener sobrepeso y obesidad. Aunque al contrario, también hay individuos que son extremadamente delgados.
- Depresión, que puede llevar al aislamiento.
- Bajo tono muscular y pobre desarrollo muscular con poca resistencia y fuerza.
Un volumen de los testículos inferior al normal.
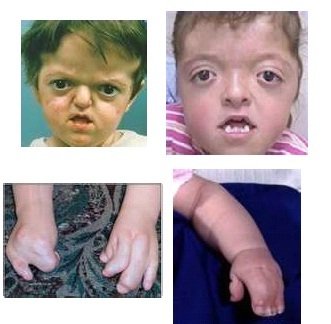

- Síndrome de Apert
El Síndrome de Apert es una afectación congénita, caracterizada por anomalías craneofaciales y de las extremidades.
Se produce el crecimiento anormal de distintos huesos en el cuerpo, principalmente en el cráneo, el área centrofacial, las manos y los pies. Su nombre se debe al físico francés E. Apert, que fue el primero en describirlo en 1906.
Normalmente, el cráneo está severamente afectado, al igual que todo el rostro, principalmente los ojos y la mandíbula. Los huesos del cráneo se unen prematuramente, evitando así el desarrollo normal del cerebro. El área centrofacial (parte del rostro a partir de la mitad de la cuenca del ojo hacia la parte superior de la mandíbula) se ve hundida, los ojos aparecen sobresalientes y los párpados caídos.
También es común la existencia de acné en el rostro, el cabello rebelde, el paladar hundido y sordera. Además, los pacientes también presentan sindactilia, fusión de los dedos de las manos y de los pies.
Muchos niños con el Síndrome de Apert también sufren de discapacidad intelectual que puede variar de leve a grave. Su lenguaje puede estar afectado y también pueden presentar problemas de comportamiento.
https://iacapap.org/content/uploads/C.1-Discapacidad-Intelectual-SPANISH-2018.pdf
https://sid.usal.es/idocs/F8/FDO25588/trastornos_desarrollo_con_discapa_intelec.pdf
